|
细菌全基因组完成图测序
细菌完成图测序是指对基因组序列未知或无近缘物种基因组信息的某个物种,构建不同插入片段的基因组文库,基于 PacBio第三代单分子测序技术结合第二代高通量测序技术,对这些文库进行序列测定,利用生物信息学方法进行拼接,获得完整的染色体及质粒序列,解析编码信息和表观遗传修饰信息。
应用领域
l 获得完整的染色体序列图谱
l 获得完整的质粒或 BAC 克隆子序列图谱
l 解析甲基化修饰相关信息(6mA和4mC)
测序优势
(1) 项目周期短:完成图最短项目周期 15 工作日,平均项目周期 30 工作日;
(2) 方案设计合理:完成图的方案以三代平台为主、二代平台为辅,平台完美结合,得到完整的染色体和质粒序列图谱;
(3) 免费赠送待测菌株的菌种鉴定,以防菌株错测;
(4) 拼接结果准确:三轮校正(拼接前利用三代测序数据间的 Overlap 对测序数据进行校正;拼接后,利用三代测序数据对拼接结果进行第二轮校正;最后,利用高质量的二代测序数据对拼接结果进行多次校正)使得到的基因组拼接结果更加准确、可靠;
(5) 项目经验丰富:细菌完成图已完成样品数量近 300 例;
(6) 细节服务到位:无论是圈图起始位点的调整,还是起始密码子的翻译(所有起始密码子的氨基酸均为甲硫氨酸),力求做到最好。
送样要求
|
送样形式 |
送样要求 |
|
菌体 |
菌体湿重 ≥ 3 g |
|
DNA |
总量 ≥ 20 µg(荧光定量);
OD260/OD280 = 1.8~2.0;
OD260/OD230 = 2.0~2.2 |
信息分析内容
|
序号 |
分析项目 |
类别 |
分析 |
备注 |
|
1 |
数据整理 |
A |
|
|
|
2 |
数据质控 |
A |
|
|
|
3 |
高质量数据获取 |
A |
|
|
|
4 |
基因组序列拼装与分析 |
A |
|
|
|
5 |
蛋白编码基因预测 |
A |
|
|
|
6 |
非编码 RNA 预测 |
A |
|
|
|
7 |
其他非编码 RNA 预测 |
A |
|
|
|
8 |
CRISPRs 预测 |
A |
|
|
|
9 |
原噬菌体预测 |
B |
|
|
|
10 |
基因岛预测 |
B |
|
完成图 |
|
11 |
致病菌毒力因子(VFDB)分析 |
B |
|
|
|
12 |
抗生素抗性(CARD)分析 |
B |
|
|
|
13 |
碳水化合物活性酶(CAZy)分析 |
B |
|
|
|
14 |
信号肽预测 |
B |
|
|
|
15 |
跨膜螺旋预测 |
B |
|
|
|
16 |
蛋白编码基因的序列比对 |
A |
|
|
|
17 |
蛋白编码基因的 GO 注释 |
A |
|
|
|
18 |
蛋白编码基因的 eggNOG 注释 |
A |
|
|
|
19 |
蛋白编码基因的 KEGG 注释 |
A |
|
|
|
20 |
蛋白编码基因的 Swiss-Prot 注释 |
A |
|
|
|
21 |
蛋白编码基因的 TrEMBL 注释 |
C |
|
|
|
22 |
蛋白编码基因的 Pfam 注释 |
C |
|
|
|
23 |
蛋白编码基因的 CDD 注释 |
C |
|
|
|
24 |
蛋白编码基因的 InterProscan 注释 |
C |
|
|
|
25 |
基因组圈图绘制 |
B |
|
完成图 |
|
26 |
基因组基本信息比较分析 |
C |
|
参考基因组数量≥1 |
|
27 |
泛基因组分析 |
C |
|
参考基因组数量≥10 |
|
28 |
基因家族分析 |
C |
|
参考基因组数量≥1 |
|
29 |
基于 16s rDNA 的系统发育树重构 |
C |
|
参考基因组数量≥2 |
|
30 |
基于单拷贝基因的系统发育树重构 |
C |
|
参考基因组数量≥2 |
|
31 |
基于 Mauve 的全基因组序列比对 |
C |
|
参考基因组数量≥1 |
|
32 |
基于 MUMmer 的全基因组序列比对 |
C |
|
参考基因组数量≥1 |
|
33 |
基因组数据上传 |
C |
|
提供相关数据 |
A+B: 完成图标准分析内容;
C: 个性化分析内容。
信息分析流程
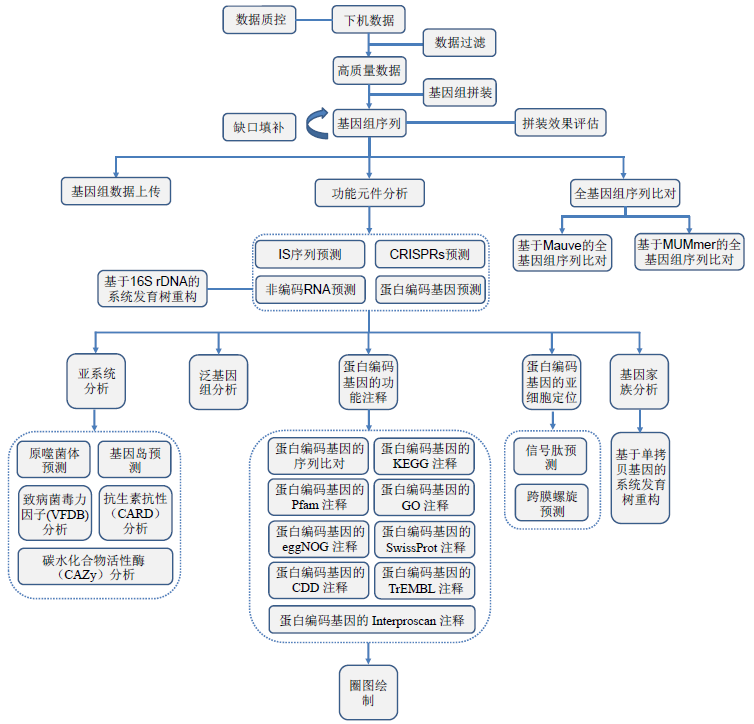
高级信息分析内容示例

图 1:Pan-Core 基因稀释曲线

图 2 :共线性分析
经典案例:
利用第三代单分子测序技术解析大肠杆菌MRE600的基因组和甲基化组
Genome Biol Evol 2016 IF:4.229
关键词:大肠杆菌;志贺氏菌;表观遗传修饰;大肠杆菌素
研究背景
大肠杆菌是一种革兰氏阴性、短杆状、能运动、无芽胞的兼性厌氧菌,通常定殖于人和动物肠道中。大部分大肠杆菌无致病性,与机体是互利共生的关系;当机体免疫力降低、肠道长期缺乏刺激等情况下,部分大肠杆菌会移居到肠道以外的地方,造成相应部位的感染或全身散播性感染。因此,大肠杆菌通常被看作机会致病菌。大肠杆菌构造简单,遗传背景清晰,培养操作容易,因此常被作为基因工程的对象加以利用。大肠杆菌 MRE600 是一株 RNAse I 活性缺失的菌株, 常被用于 mRNA、tRNA、rRNA 的表达和纯化。本文采用 PacBio 平台获得了 MRE600 的全基因组序列图谱和甲基化图谱。
实验方法
供试样本:大肠杆菌 MRE600 菌株;
文库大小:20,000 bp;
测序试剂: P5-C3;
测序平台:PacBio RS II;
研究结果
采用第三代单分子测序对大肠杆菌 MRE600 进行全基因组从头测序,总共获得 1 条染色体序列(4.83 Mb)和 3 个质粒(89.1 kb、56.9 kb和7.1 kb)。基因组的 G+C 含量为 50.8%,包含 5,181 个基因,其中,蛋白编码基因为 4,913 个,RNA 基因为 268 个,41,469 个碱基发生修饰(0.83%)。
图 1 大肠杆菌MRE600染色体和质粒组装结果:细菌完成图参考文献1中的fig 1.
表 1 大肠杆菌MRE600的表观基因组检测:细菌完成图参考文献中的表3

对 RNAse I 的编码基因 rna 进行分析发现,该基因的第五个密码子为提前终止的密码子,即 MRE600 不能产生全长 RNase I 蛋白,这也解释了 MRE600 的 RNase I 活性丧失的原因。
参考文献
Kurylo CM, et al. Genome Sequence and Analysis of Escherichia coli MRE600, a
Colicinogenic, Nonmotile Strain that Lacks RNase I and the Type I Methyltransferase, EcoKI. Genome Biology and Evolution, 2016, 8 (3): 742-752.
项目经验
|
物种拉丁名 |
基因组大小(Mb) |
GC含量(%) |
派森诺已完成数目 |
|
Acinetobacter junii |
3.37 |
38.7 |
6 |
|
Bacillus cereus |
5.64 |
35.2 |
8 |
|
Bacillus pumilus |
3.72 |
41.4 |
15 |
|
Bacillus.mycoides |
5.72 |
35.3 |
9 |
|
Bacillus.sp |
4.89 |
40 |
24 |
|
citrobacter freundii |
5.26 |
51.6 |
15 |
|
clostridium tetani |
2.85 |
28.5 |
45 |
|
enterococcus faecalis |
3.03 |
37.3 |
6 |
|
lactobacillus crispatus |
2.2 |
36.85 |
9 |
|
Mycobacterium abscessus |
5.13 |
64.1 |
12 |
|
Neisseria meningitis A |
2.16 |
51.7 |
9 |
|
pseudonocardia |
6.55 |
73.48 |
6 |
|
Staphylococcus aureus |
2.85 |
32.8 |
60 |
|
stenotrophomonas rhizophila |
4.64 |
668.8 |
15 |
|
streptococcus suis |
2.07 |
41.2 |
30 |
|
streptomyces cuspidosporus |
8.2 |
71.9 |
3 |
|
streptomyces scabies |
9.9 |
71.25 | |